结直肠癌(CRC)每年在全球造成近500,000人死亡。尽管CRC主要与老年和不良饮食习惯有关,但导致CRC发展的确切病理生理机制仍然难以捉摸。
现在,由日本东京理科大学YoichiroIwakura教授和中国中山大学CeTang教授领导的研究小组最近能够使用小鼠模型和临床样本确定潜在机制。他们的结果已发表在NatureCommunications上。
“我们通过分析小鼠肠道肿瘤模型和来自CRC患者的临床样本,研究了Dectin-1在结直肠肿瘤发生中的作用。我们发现Dectin-1信号通过增强前列腺素E2(PGE2)的产生来促进结直肠肿瘤的发展,它通过抑制肿瘤抑制性IL-22结合蛋白(IL-22BP)的表达来促进CRC的发展,”Iwakura教授说。
Dectin-1主要用作受体蛋白,并优先结合β-葡聚糖——天然存在于各种真菌细胞壁中的葡萄糖聚合物。尽管之前的研究表明Dectin-1可以防止真菌入侵,但目前的研究强调了它作为参与CRC发展的受体蛋白的作用。
为了充分了解Dectin-1在CRC中的病理生理作用的潜在机制,研究团队培育出了Dectin-1缺陷的转基因“Clec7a–/–小鼠”。为此,该团队使用了人类家族性腺瘤性息肉病的ApcMin小鼠模型(一种以多发肿瘤为特征的癌症),以及氧化偶氮甲烷(AOM)-葡聚糖硫酸钠(DSS)诱导的结直肠癌化学致癌模型。
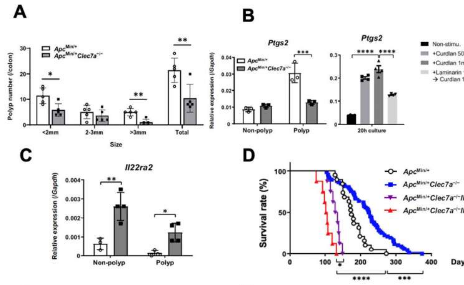
非常有趣的是,Clec7a–/–小鼠在上述两种模型中均显示肿瘤发生减少,从而强调了Dectin-1在CRC发展中的作用。
接下来,研究人员决定研究肠道细菌在肠道肿瘤发生中的作用。为此,他们创造了无菌(GF)小鼠,它们的肠道中没有共生细菌。他们发现,在完全没有任何肠道细菌的情况下,与野生型GF小鼠相比,Clec7a−/−GF小鼠的结直肠息肉数量大大减少,表明肠道微生物群不参与Clec7a息肉的减少–/–老鼠。
该团队随后决定深入研究相关的作用机制。随后基于小鼠模型的实验表明,Clec7a-/-小鼠肿瘤中的PGE2水平降低。此外,他们还观察到PGE2合酶(例如已知可促进肠道肿瘤发生的COX2)的表达减少。
此外,在研究产生PGE2合酶的细胞类型时,研究人员发现它主要是由已经浸润到结直肠肿瘤中的骨髓细胞来源的抑制细胞(MDSCs)产生的。此外,研究人员还证明PGE2促进了MDSCs的分化和增殖,进一步促进了小鼠模型中CRC的发展。
在试图阐明潜在的作用机制的同时,研究人员还注意到Clec7a-/-小鼠的IL-22BP产量增加——一种可以通过结合和抑制促炎蛋白白细胞介素来抑制结直肠肿瘤发展的蛋白质——22(IL-22)。删除负责IL-22BP表达的基因导致ApcMin小鼠的息肉增加和早期死亡,从而强调了IL-22BP在肿瘤抑制中的作用。此外,发现IL-22BP的产生被PGE2强烈抑制。
有趣的是,昆布多糖(一种来自海藻的低分子量β-葡聚糖)在喂食该化合物的小鼠中显着抑制了AOM-DSS诱导的结肠肿瘤发生。该团队还发现,虽然高分子量β-葡聚糖促进肿瘤生长,但低分子量β-葡聚糖通过抑制Dectin-1信号传导来抑制它。
这些结果也具有直接的临床意义。例如,研究小组注意到,与CLEC7A高表达(在MDSCs中)相比,具有低CLEC7A表达的CRC患者存活时间更长。此外,在CRC患者中,与正常组织相比,肿瘤中IL22RA2的表达降低,而PTGS2(一种PGE2合成酶)的表达增加。
Iwakura教授总结道:“Dectin-1通过改变PGE2和IL-22BP水平,在小鼠和人类结肠直肠肿瘤发生的发展中起着关键作用。因此,Dectin-1是一个有吸引力的靶标开发新型抗CRC疗法。”